
Benndorf Group, Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg, Service Center: Bielefeld-Giessen Resource Center for Microbial Bioinformatics - BiGi
Proteomics is a field of molecular biology that targets improvements in medical, pharmaceutical and environmental research, and diagnostics. The computational analysis of the recorded mass spectrometry data has become one of the biggest challenges in the field of proteomics, in particular the problem of determining the validity of an identified peptide-spectrum match (PSM). Metaproteomics, which applies proteomics tools to microbial communities, faces further challenges regarding validation of PSMs.
Commonly employed methods for the validation of PSMs, like an FDR estimation with the Target-Decoy approach, rely heavily on expert systems and require the entirety of the data set at once to work. These methods are highly dependent on carefully designed scoring functions that use varying feature representations to achieve validation. Multiple issues arise, such as different outputs depending on method used and the inapplicability for streaming. In metaproteomics, protein database choice is an even bigger issue for PSM validation, because unlike in single-organism proteomics, protein databases are large to accommodate the uncertainty about the species present in a given sample, which in turn negatively influences PSM validation.
Our new implementation - Deep-learning Peptide Spectrum Identification Validator (DeePSIVal) - employs a deep learning architecture comprised of multiple GRU units organized in a many-to-one configuration to generate a multi-dimensional representation of the pairs of peptide sequences and spectra. A CNN union model interprets the tensor representation to correctly validate the encoded peptide spectrum match. Therefore, DeePSIVal relies on raw data and automatic feature detection. By using this method, PSM validation is independent of the protein database used, thus eliminating the effects of database size.
In our previous work, a machine learning approach was implemented and trained with prepared mass spectrometry data sets done for this specific purpose as well as data sets from the public database PRIDE. We will compare the DeePSIVal against other approaches with regard to model performance and time performance.
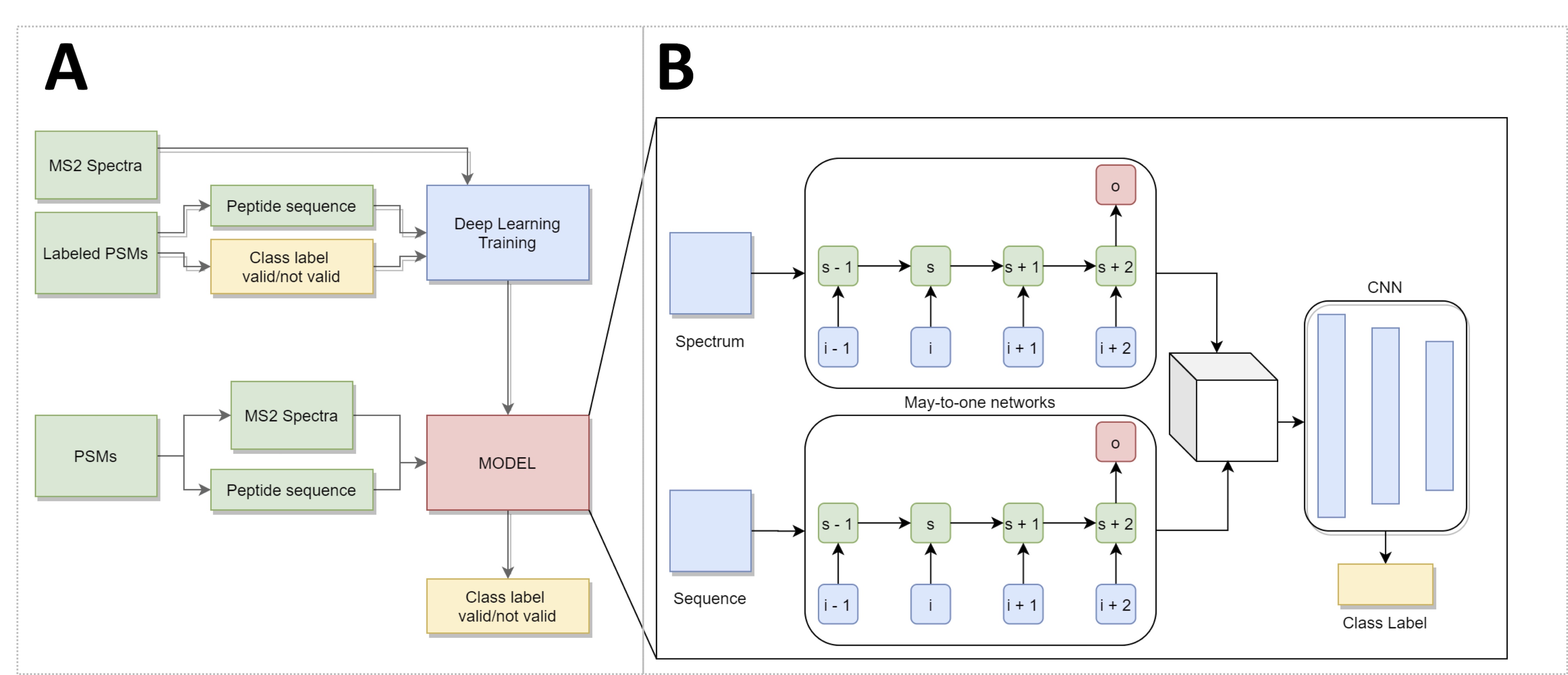
Workflow (A) and architecture (B) of the implemented DeePSIVal prototype. (A) MS2 spectra and the peptide sequence – the raw data for a peptide spectrum match – are used directly as input. For training a Class label has to be provided; the finished model can output a class label for new PSMs. (B) Deep learning architecture consists of 3 components. Two Many-to-One networks read the spectrum and sequence data of a PSM. The resulting output is concatenated and evaluated by a convolutional neural network, which will convert it into a class label.
Search projects by keywords: