-
Cardiovascular risk modelling
Eils Group, Hub for Innovations in Digital Health, Service center: Heidelberg Center for Human Bioinformatics – HD-HuB
In this project, we work towards an improved prevention and treatment of patients with established coronary heart disease (CHD). CHD is a major cause of morbidity and mortality. The risk landscape in patients with coronary disease is highly heterogenous, depending on their genetic background, clinical characteristics, cardiovascular risk factors and atherosclerotic disease status. With the availability of effective, but costly novel treatment options (such as PCSK9-inhibitors), there is great need for advanced cardiovascular risk prediction tools to stratify patients, guide the use of novel treatments and improve clinical trial design by selecting high-risk individuals. Here, we aim to improve and personalize prevention and treatment of coronary heart disease by developing data-driven, neural-network based tools for risk modelling and multi-modal data integration. We investigate representation learning techniques to identify latent factors across data modalities associated with risk and examine lifetime risk more closely with the aim of making individualized risk prediction using counterfactuals. The clinical environment at Charité in synergy with data-collection and patient management platforms allows for a prospective validation and clinical integration of our technologies.
For further information, visit ails lab.
 Search projects by keywords:
Search projects by keywords: -
Competing Endogenous RNA Networks in Colorectal Cancer
Stadler Group, Leipzig University, Service Center: RNA Bioinformatics Center - RBC
Colorectal cancer (CRC) is a heterogeneous cancer and its treatment can be different depending on its anatomical location. Some molecules can act as regulatory network influencing different aspects of cancer development. This study aimes to create competing endogenous RNA (ceRNA) networks to find biomarkers that elucidate clinical and functional implications for colon, rectum and rectosigmoid junction cancer via means of machine learning, based on RNA expression and clinical data of patients obtained from The Cancer Genome Atlas (TCGA).
For more information see RNA Bioinformatics Center (RBC)
Funded by: CAPES (Brazil)
Search projects by keywords:
-
Deep Learning for Analyzing Microscopy Images and Cellular Phenotyping
Rohr Group, Heidelberg University, Service center: Heidelberg Center for Human Bioinformatics – HD-HuB
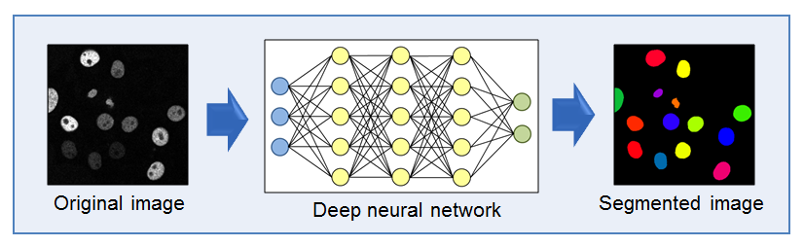
Automated analysis of high-throughput and high-content microscopy image data is important to elucidate biological processes. However, analyzing such data poses a number of challenges. Recently, deep learning methods within the field of artificial intelligence emerged which yield superior results compared to classical image analysis methods.
The Biomedical Computer Vision Group at Heidelberg University is developing deep learning methods for computer-based analysis of cell microscopy image data (see de.NBI Services). The methods are based on deep neural networks that are trained from example data. Convolutional neural networks and recurrent neural networks are developed for different image analysis tasks such as cell segmentation and classification of microscopy images. The aim is to improve the automated quantification of cellular phenotypes at the single cell level as well as to efficiently process large scale microscopy data.
For further information, visit the websites of the Biomedical Computer Vision Group or HD-HuB.
Search projects by keywords:
-
Identification of new antimicrobial resistance targets by high-throughput deep learning
Goesmann Group, Gießen University, Service Center: Bielefeld-Giessen Resource Center for Microbial Bioinformatics - BiGi
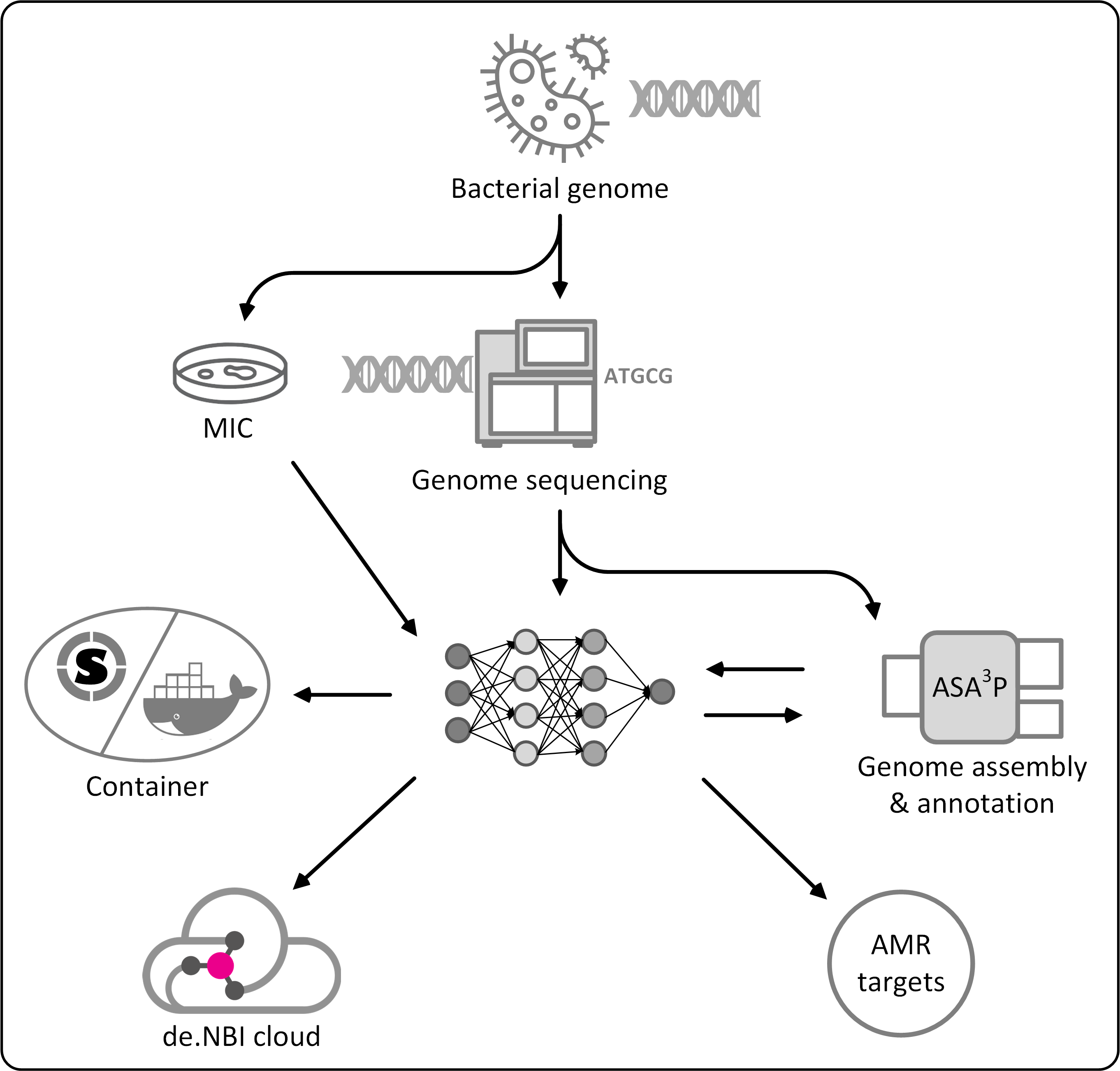
Antibiotic resistant bacteria have become a global severe threat for public health. Their surveillance and containment as well as the timely identification of new genetic determinants of antibiotic resistances are important tools to fight back emergent as well as established pathogens. Addressing these challenges, researchers from universities in Giessen and Marburg have started a new project called Deep-iAMR to combine expertise in medical microbiology, machine learning and cloud computing techniques. The aim of this new collaboration is to screen large amounts of genomic as well as phenotypic data for complex patterns between genomic and phenotypic data to find the genetic determinants of hard to detect or entirely new antimicrobial resistances. However, the application of modern artificial intelligence methodologies to find these patterns requires massive computing resources as well as specialized hardware, such as GPUs, tensors and FPGUs.
Researchers within the Deep-iAMR project take advantage of the cloud computing infrastructure of the de.NBI network which provides required computing resources as well as specialized hardware on demand. Resulting new methodologies and scalable tools will be shared with the scientific community and discovered new antibiotic targets will help to design necessary and urgently required new drugs to cure infections with highly-resistant pathogens.
For further information, please visit Deep-iAMR. To access the de.NBI Cloud visit de.NBI Cloud.
 Search projects by keywords:
Search projects by keywords: -
LAMarCK - LongitudinAI Multiomics Characterization of disease using prior Knowledge
Zeller Group, Saez-Rodriguez Group, Schlesner Group, Bork Group, and Korbel Group, Service center: Heidelberg Center for Human Bioinformatics – HD-HuB
The need for proper integration of molecular omics data from human individuals is arguably one of the biggest challenges in current biomedical research, as these data cover different functional aspects that facilitate basic research, but also reveal differences between human physiological states with medical relevance (e.g. disease versus healthy, different disease subtypes etc.). To tackle this challenge, we formed a team of researchers with complementary areas of expertise from different Heidelberg institutions, all associated with the de.NBI node HD-HuB, which delivers services on human genomics and human microbiomics. Both these fields rely on similar kinds of data, which are increasingly coming from longitudinal studies assessing multiple omics approaches. Thus, they face similar technical and methodological challenges - and closer communication, as well as integrative approaches bridging both fields hold great promise for improved diagnostic, prognostic and therapeutic approaches, as is already becoming apparent for type-2-diabetes onset.
The LAMarCK consortium on the one hand aims to devise generic approaches to integrate heterogeneous longitudinal multi-omics data across microbiome and host, on the other hand it will also develop frameworks that make use of domain-specific prior knowledge on the underlying biological mechanisms for improved interpretability of the results. Funded by the BMBF (grant no. 031L0181A), the consortium is applying and benchmarking state-of-the-art machine learning approaches for longitudinal multi-omics data from microbial and host cells and augmenting these by integration with biomolecular knowledge bases. These new approaches will help dissect the molecular mechanisms underlying human diseases, such as colorectal cancer.
Search projects by Keywords:
-
Omni-genetic phenotype models
Eils Group, Hub for Innovations in Digital Health, Service center: Heidelberg Center for Human Bioinformatics – HD-HuB
Genetic contributions to many health-related phenotypes like blood pressure and body mass index often arise from complex interactions between multiple genes. Thus, an understanding of how variants of different genes interact, is of great interest. We plan to make use of recent advances in the field of explainable machine learning to analyze data from large cohort studies. Starting with variants of individual genes, we want to model how they affect biological systems, for example DNA repair. Based on the combination of smaller specific subsystems, we want to predict the activity of more complex systems and repeat this process until the information of all genes is combined in one system describing the effect of all variants. At that point, the information on thousands of genes will be available in a very compressed representation, which will be the basis to predict phenotypes like blood pressure or other medically relevant phenotypes.
Additionally, this approach can be used to investigate which cellular systems in the model are affected by a given set of variants resulting in a specific phenotype. For example, for an individual with increased blood pressure and variants in multiple genes, this could be used to predict, whether the DNA repair system is involved in the increased blood pressure. Information like this can be used to check if the model makes its predictions in an intuitive way and can also be the starting point of new research, if strong connections are found.
For further information, please visit ails lab.

-
REmatch - Cloud AI for drug discovery
Boutros Group, DKFZ Heidelberg, Service center: Heidelberg Center for Human Bioinformatics – HD-HuB
In the REmatch project we develop cloud technologies that combine advanced image analysis and an artificial intelligence (AI) to predict a drug’s mechanisms of action and its polypharmacology. By this means, REmatch aims to save resources, to avoid unnecessary complex tests, and to facilitate drug repurposing by identifying new applications for approved drugs.
High drug attrition rates, few discoveries of new drug targets and patent cliff together pose a major problem for drug development. Despite better technologies and a deeper understanding of biological mechanisms, the efficiency at which novel drugs reach the market steadily declines. Many candidates fail late in clinical development due to side-effects or the lack of a therapeutic window. In addition, many promising or approved drugs harbour unrecognized therapeutic efficacy in other indications that were not in the focus during initial development.
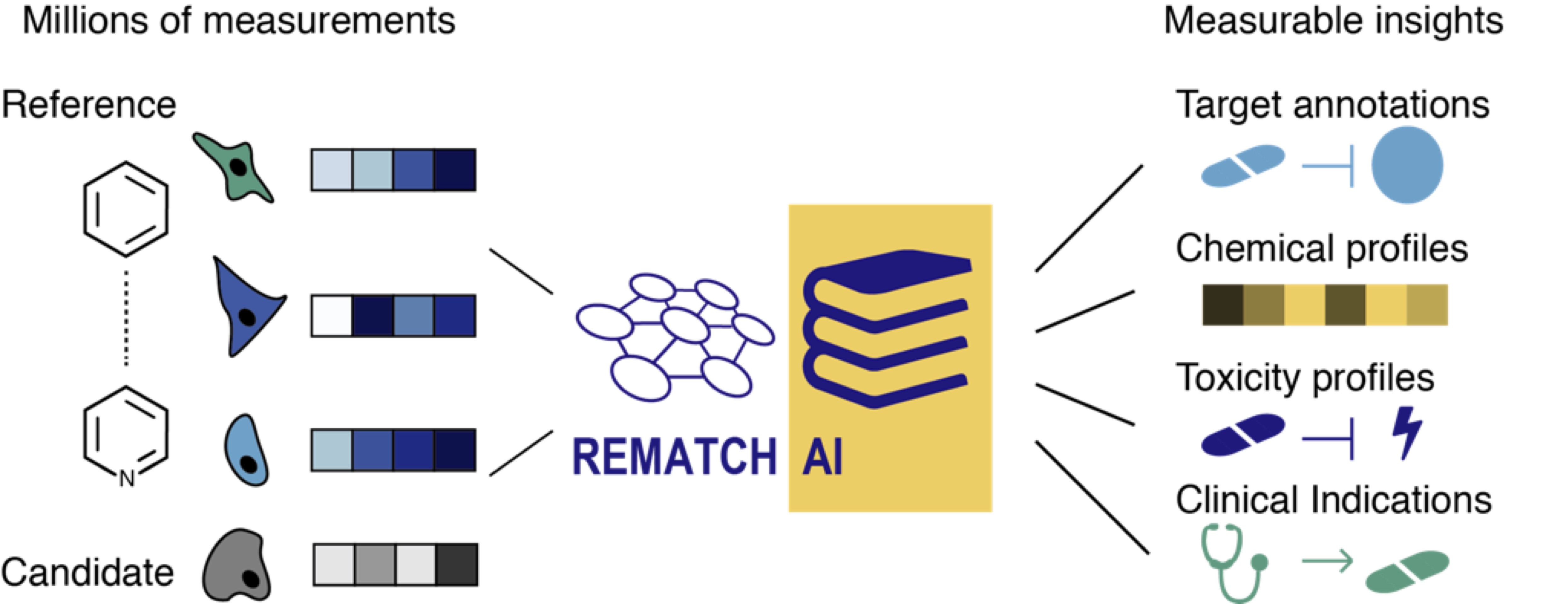
As part of the REmatch project, we develop machine learning strategies to compare drug responses of candidate drugs to a reference database containing the responses of thousands of approved drugs. This facilitates the profiling of side-effects such as toxicity for candidate drugs. By comparing the responses of known drugs, REmatch further allows to identify drugs which harbour potential for other indications than they were initially developed for. In our approach, the responses of candidates and known drugs are represented as profiles which are derived from images of cultured cells that were exposed to these drugs. We generate such images at low costs and in a highly parallel fashion using an automated microscopy platform. REmatch can be used early during the drug discovery process to flag candidates sharing e.g. known toxicity of marketed drugs. REmatch employs a cloud-based, scalable end-to-end image analysis to derive phenotypic profiles of drug treated cells in a highly efficient manner. This removes a major bottleneck arising when thousands to millions of images are collected. With REmatch we offer a solution that enables researchers to fast and cost-efficiently characterize candidate molecules during pre-clinical development.
REmatch is supported by an ERC Proof of Concept grant and as a part of the proof-of-concept evaluation we used the de.NBI Cloud to build a reference database that contains the responses of more than 2000 drugs in 20 cancer cell lines. The cloud resources were used to streamline the analysis of hundreds of thousands of images with the REmatch technology. REmatch delivers our analysis and data processing pipelines, as well as access to the reference database in any cloud. The concept of cloud-based analysis, know-how and infrastructure addresses an unmet need and makes a routinely employment of image-based profiling analysis in drug discovery commercially attractive for pharmaceutical companies and viable for academic labs.
For further information, visit the website of the Division of Signaling and Functional Genomics at DKFZ Heidelberg.
Search projects by keywords:
